Dravethon 2026
Ensemble pour les personnes touchées par le syndrome de Dravet Du 1er au 23 juin 2026, c’est le retour du Dravethon ...
Ensemble pour les personnes touchées par le syndrome de Dravet Du 1er au 23 juin 2026, c’est le retour du Dravethon ...
C’est à Nancy à la salle des fêtes de Gentilly que nous avons Sylvia et moi-même Dominique, référentes régionales Grand ...
Encore une année qui s’achève, la 16ème depuis l’existence de l’Alliance Syndrome de Dravet. L’association continue sans relâche de soutenir ...
Vous avez surement lu dans notre dernière newsletter ces mots de Maher, notre président : « A l’heure où j’écris ces ...
«Voilà que l’été arrive. Et c’est le moment de notre magazine annuel. Et oui, notre newsletter a pris une autre ...
En mars et en avril 2025, ASD a proposé aux familles de personnes atteintes du syndrome de Dravet deux weekends ...
La Journée Internationale des Maladies Rares (JIMR) a eu lieu ce vendredi 28 février.Le syndrome de Dravet est considéré comme ...
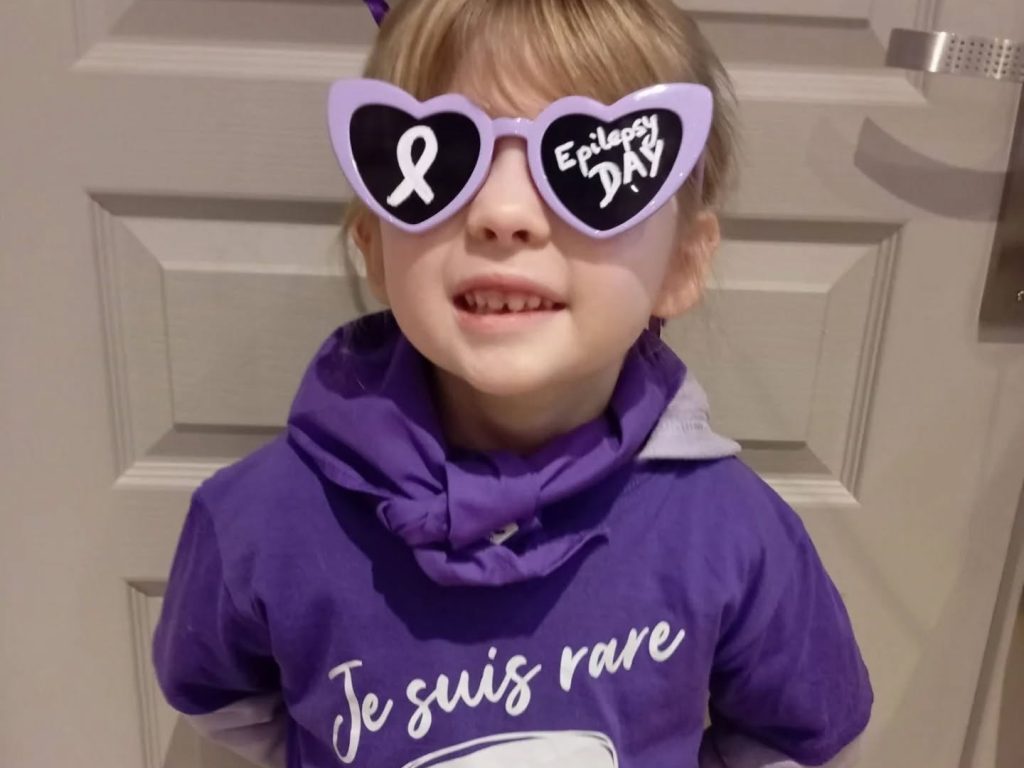
La journée internationale de l’épilepsie (JIE) a eu lieu ce lundi 10 février 2025, vous avez été nombreux à participer ...
Porter la voix des familles de personnes atteintes du syndrome de Dravet est un des axes de travail d’ASD dans ...
Cette année encore les rencontres annuelle ont rassemblé plus d’une centaine de participants à l’Espace Batignolles à Paris ainsi qu’une ...