Journées recherches de DéfiScience 15 et 16 mai 2025
Tout comme en 2023, la filière de santé maladies rares DéfiScience a organisé à Paris, le 15 et 16 mai ...
Tout comme en 2023, la filière de santé maladies rares DéfiScience a organisé à Paris, le 15 et 16 mai ...
En mars et en avril 2025, ASD a proposé aux familles de personnes atteintes du syndrome de Dravet deux weekends ...
Voici une nouvelle présentation d’un article scientifique qui peut vous intéresser.Si vous souhaitez creuser le sujet, n’hésitez pas chercher l’article ...
La Journée Internationale des Maladies Rares (JIMR) a eu lieu ce vendredi 28 février.Le syndrome de Dravet est considéré comme ...
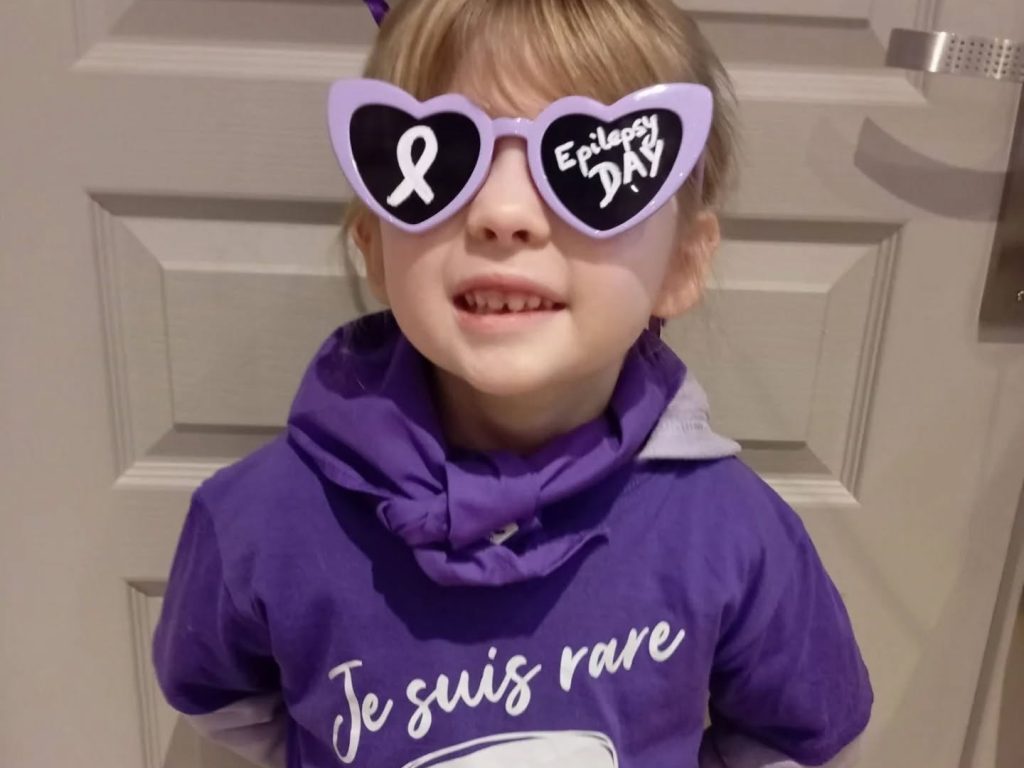
La journée internationale de l’épilepsie (JIE) a eu lieu ce lundi 10 février 2025, vous avez été nombreux à participer ...
Cette année encore les rencontres annuelle ont rassemblé plus d’une centaine de participants à l’Espace Batignolles à Paris ainsi qu’une ...
Laurie, Rebecca et Manue ont représenté ASD lors du congrès de la Société Française de Neurologie Pédiatrique à Lyon du ...
Voici une nouvelle présentation d’un article scientifique qui peut vous intéresser.Si vous souhaitez creuser le sujet, n’hésitez pas chercher l’article ...
Voici une nouvelle présentation d’un article qui peut vous intéresser.Si vous souhaitez creuser le sujet, n’hésitez pas chercher l’article complet.Ce ...